| Investigations of Protein stability and Folding | |||||||||||||||||||||||||||
|
The rate of expansion of information technology, its
associated use in depositing and accessing large number of biological
data has created unprecedented need for researchers and R&D
industry. Computational tools developed in conjunction with the wet-lab
techniques generate high levels of data as output. However these outputs
are massive and the computed biological information (bioinformatics)
provides us the ways and means to correlate these massive data with
knowledge. In this direction, our work addresses important problems in
molecular biology, like protein stability and folding. A series of
computational analyses were performed to elucidate the factors
responsible for it. Eventhough the experimental analyses of these
studies have provided a wealth of information, the computational
approaches has provided the key results to perform protein engineering
experiments. The most promising methods looking ahead are those that
systematically control false positive production rate by integrating
computation with information obtained from different experimental
angles. Further there is a place for computational analysis to augment
nascent experimental methods in evolving disciplines of functional
genomics and proteomics. In line with this, a study was carried out on single mutants of T4 and human lysozymes using free energies and amino acid properties which demonstrate the importance of classifying them based on secondary structure towards the stability. Further the significance of secondary structural elements are identified from the study of 1531 single mutants using hydrophobic profile, long-range order, stabilization centers and amino acid conservation. In a comparative analysis of thermophilic and its homologous mesophilic proteins, we inferred the importance of hydrophobic free energy. We have also analysed the stability upon buried, partially buried and surface mutations based on different secondary structural elements using various amino acid properties, and also proposed a method to predict the stability of protein mutants. In the analysis of folding rate of two-state proteins, the significance of various free energies of the non-covalent interactions, amino acid properties and topological parameters are explored. |
|||||||||||||||||||||||||||
| Role of amino acid properties to determine backbone tau(N-Calpha-C') stretching angle in peptides and proteins | |||||||||||||||||||||||||||
| The analysis of the basic geometry of amino acid residues of protein structures has demonstrated the invariability of all the bond lengths and bond angles except for tau, the backbone N-Calpha-C' angle. This angle can be widened or contracted significantly from the tetrahedral geometry to accommodate various other strains in the structure. In order to accurately determine the cause for this deviation, a survey is made for the t angles using the peptide structures and the ultrahigh resolution protein structures. The average deviation of N-Calpha-C' angles from tetrahedral geometry for each amino acid in all the categories were calculated and then correlated with forty-eight physiochemical, energetic and conformational properties of amino acids. Linear and multiple regression analysis were carried out between the amino acid deviation and the 48 properties. This study confirms the deviation of tau angles in both the peptide and protein structures but similar forces do not influence them. The peptide structures are influenced by physical properties whereas as expected the conformational properties influence the protein structures. And it is not any single property that dominates the deviation but the combination of different factors contributes to the tau angle deviation. | |||||||||||||||||||||||||||
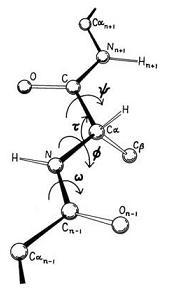 |
|||||||||||||||||||||||||||
| Study of weak interactions in a few heterocyclic structures | |||||||||||||||||||||||||||
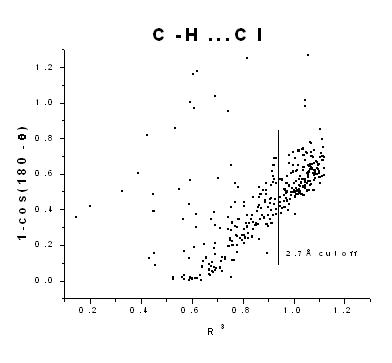
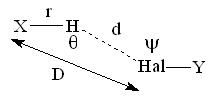
| The hydrogen bonding nature of the halogen atom acceptors are analyzed statistically and the geometrical characteristics of this weak hydrogen bonded interactions are studied for the oxygen, nitrogen and carbon atom donors. The hybridization states of the donor atoms and the different coordination environment of the halogen acceptors were also considered. The distance and the directionality characteristic that dictates the hydrogen bonds are given importance. Oxygen and nitrogen donors showed similar characteristics of a strong hydrogen bond whereas carbon donors had weak nature. The hydrogen bonding nature of the carbon donor with halogen acceptor was proved from this analysis but within a hydrogen…acceptor distance cutoff of 2.7Å. Different hybridization state of the donor atoms did not produce significant deviation in the interacting nature but the various coordination environments did show some deviation. | |||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||
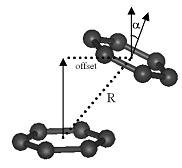
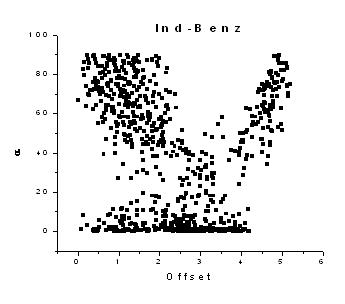
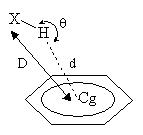
| Non-covalent interactions involving heteroaromatic ring systems play a major role in determining the function of many chemical as well as biological molecules. Therefore, detailed quantitative analyses of the p-interactions (X-H…pi and pi…pi) were carried out for nitrogen containing p-systems (isoxazole, imidazole and indole moieties). Statistical analysis of the geometrical properties for the oxygen, nitrogen and carbon atom donors with the heterocyclic p-system acceptors showed that carbon donors participate relatively in large numbers for X-H…pi interactions and they adopt T-shaped geometry. The pi…pi interaction analysis was categorized into three types based on the involvement of the heterocyclic pi-systems with themselves, with any other benzene ring present in that particular structure and their influence on the formation of pi-interactions between benzene-benzene rings found in that particular compound. The pi-systems in all the three categories prefer to form offset stacking pi…pi interaction geometry. The benzene rings which normally favour the formation of T-shaped geometry is found to prefer offset stacking geometry which may be due to the influence of the heteroatom. The pi-systems in these heterocyclic structures behaves similar to the Phenylalanine - Phenylalanine interactions in proteins and therefore this quantitative analysis can serve as a guide for structural and biological studies. | |||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||
| Average Assignment Method for Predicting the Stability of Protein Mutants | |||||||||||||||||||||||||||
| Prediction of protein stability upon amino acid substitutions is an important problem in molecular biology and it will be helpful for designing stable mutants. In this work, we have analyzed the stability of protein mutants using three different data sets of 1791, 1396 and 2204 mutants, respectively for thermal stability (DTm), free energy change due to thermal (DDG) and denaturant denaturations (DDGH2O), obtained from ProTherm database. We have classified the mutants into 380 possible substitutions and assigned the stability of each mutant using the information obtained with similar type of mutations. We observed that this assignment could distinguish the stabilizing and destabilizing mutants to an accuracy of 70-80% at different measures of stability. Further, we have classified the mutants based on secondary structure and solvent accessibility and observed that the classification significantly improved the accuracy of prediction. The classification of mutants based on helix, strand and coil distinguished the stabilizing/destabilizing mutants at an average accuracy of 82% and the correlation is 0.56; information about the location of residues at the interior, partially buried and surface of a protein correctly identified the stabilizing/destabilizing residues at an average accuracy of 81% and the correlation is 0.59. The nine sub-classifications based on three secondary structures and solvent accessibilities improved the accuracy of assigning stabilizing/destabilizing mutants to an accuracy of 84-89% for the three datasets. Further, the present method is able to predict the free energy change (DDG) upon mutations within the deviation of 0.64kcal/mol. We suggest that this method could be used for predicting the stability of protein mutants. | |||||||||||||||||||||||||||